Introducere. Epidemiologie
Cancerul colorectal (CCR) reprezintă o cauză importantă de morbiditate şi mortalitate, această patologie fiind la ora actuală a doua cauză de mortalitate neoplazică în România, după cancerul pulmonar. CCR reprezintă una dintre cele mai frecvente neoplazii umane, afectând o persoană din 20 în ariile cu standard socioeconomic ridicat. Reprezintă a treia cauză de cancer la ambele sexe, după plămân şi stomac la bărbaţi şi după sân şi uter la femei. Aproximativ unul din opt cancere este de origine colorectală, iar CCR este responsabil pentru aproximativ 1 din 10 decese de cauză neoplazică.
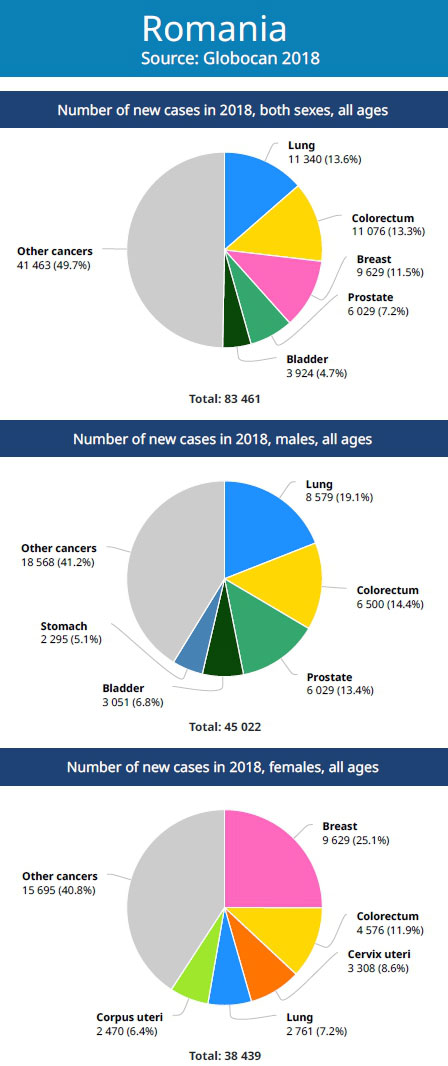
În România, 80.000 de pacienţi au fost diagnosticaţi cu cancer anul trecut şi s-au înregistrat peste 50.000 de decese. Cancerul colorectal este pe locul doi ca incidenţă şi ca mortalitate, cu peste 11.000 de cazuri noi şi peste 6000 de decese anul trecut, potrivit raportărilor oficiale.
Aproximativ 2-5% din totalitatea cazurilor de cancer colorectal apar datorită unei entități definite de sindrom neoplazic. Sindromul Lynch (cunoscut anterior ca și cancerul colorectal ereditar non-polipozic, HNPCC) și polipoza adenomatoasă familială (FAP) sunt patologii genetice autozomal dominante, reprezentând majoritatea patologiei neoplazice colorectale. Alte sindroame ale cancerului colorectal familial mai puțin frecvente includ sindroamele hamartomatoase, sindroamele de polipoză hiperplazică, sindroamele polipozice autozomal recesive asociate cu gena MYH și polipoza familială juvenilă. Deși incidența cancerelor colorectale ereditare este relativ scăzută comparativ cu incidența cancerelor colorectale cu componentă sporadică, identificarea precoce a indivizilor cu risc înalt și a familiilor lor este importantă pentru inițierea oportună a profilaxiei și înscrierea în programe de supraveghere.
Sindromul Lynch este cauzat de mutațiile germinale ale genelor de reparare a erorilor de complementaritate ADN („mismatch repair” – MMR), precum MLH1, MSH2, MSH6 și PMS2. Concomitent, rearanjamente genomice în cadrul genei EPCAM, cu rol în sintetizarea moleculei de adeziune a celulelor epiteliale, pot duce la silențierea genei MSH2, aflată în strânsă legătură cu gena EPCAM, în țesuturile ce o exprimă. Cancerele colorectale din cadrul sindromului Lynch sunt caracterizate de o rație a progresiei adenom-carcinom de 1:1 (timp estimat de transformare malignă de 1-3 ani), comparativ cu cele sporadice, cu o rație de 30:1 (timp estimat de transformare malignă de 8-17 ani). Dacă nu sunt tratați, majoritatea polipilor se vor transforma malign, așa cum s-a observat la 70% dintre pacienții în vârstă de 70 de ani și la 80% dintre pacienții cu vârsta de 85 de ani. S-a observat o incidență crescută a cancerelor colorectale metacrone și sincrone, împreună cu dezvoltarea unui cancer colorectal secundar în 30% din cazuri după 10 ani și 50% din cazuri după 15 ani. Sindromul Lynch predispune la neoplazii extracolonice, precum cele endometriale, gastrice, ovariene, intestinale, ale epiteliului hepatobiliar, epiteliului urotelial și neoplasmul cerebral.
Istoric
Istoria sindromului Lynch datează începând cu anul 1895, pe vremea când Aldred Scott Warthin lucra ca și director al Departamentului de Patologie a Școlii de Medicină din Ann Arbor, aparținând Universității Michigan. Croitoreasa sa, de origine germană, prezenta gânduri de îngrijorare și depresie despre posibilitatea de a dezvolta cancer gastric, uterin sau de colon, similar cu membrii familiei sale. Aceste gânduri s-au adeverit, aceasta decedând la o vârstă tânără din cauza unui cancer endometrial. Warthin a studiat familia sa în detaliu și a publicat în 1913 un pedigree extins ce identifica 10 membri afectați din familia sa, descriind multe generații cu patologie neoplazică a colonului, stomacului și uterului. A realizat un audit a 3600 de cazuri cu neoplazie, diagnosticate între anii 1895 și 1912, și a observat că aproximativ 15% dintre cei ce au avut un istoric familial pozitiv de carcinom au dezvoltat în cele din urmă un carcinom. Warthin a concluzionat că exista o „influență ereditară asupra cancerului”. Un raport actualizat asupra Familiei G a fost publicat în anul 1925. S-a observat o frecvența crescută a cancerelor tractului gastrointestinal și uterului. Aceste tipuri de neoplazii apăreau pentru membrii familiei la o vârstă medie de 37,9 ani, iar tendința cancerului colorectal de a se forma avea dispunere la nivelul colonului proximal. Warthin a decedat în anul 1931.
În 1966, Henry Lynch a descris două familii din statele Nebraska și Michigan ale Statelor Unite ale Americii, ce prezentau un model similar de dezvoltare a neoplaziilor, implicând multiple generații, în mod asemănător familiei G. A studiat datele provenite de la 650 de membri ai familiei G și a publicat în cele din urmă, în anul 1971, un manuscript intitulat „Cancers Family G, Revisited”, ce a solidificat dovezile ce caracterizau această entitate sindromică având un model de transmitere ereditară de tip autozomal dominant și o vârstă de debut precoce (vârstă de debut < 45 de ani), ce implica adenocarcinoame ale colonului, endometrului și stomacului. Multiple alte raportări descriind HNPCC au fost publicate la mijlocul anilor 1980, iar un număr deosebit de scheme de clasificare clinică au fost dezvoltate, cu scopul cercetării.
În anul 1989, International Collaborative Group on HNPCC (ICG-HNPCC) a fost înființat pentru a dezvolta un set de criterii, cunoscut ca și „Criteriile Amsterdam-I”, pentru a diagnostica HNPCC și pentru a facilita identificarea genelor cauzatoare. Aceste criterii au fost lărgite pentru a include tumorile extracolonice, acum cunoscute ca și „Criteriile Amsterdam-II”. Odată cu identificarea câtorva mutații în genele MMR (MLH1, MSH2, MSH6, PMS2), National Cancer Institute (NCI) a organizat un workshop internațional pentru sindromul Lynch în Bethesda, în cursul lunii noiembrie, 1997. Au prezentat un tablou standardizat de diagnosticare cu ajutorul markerilor pentru microsateliți și au dezvoltat ghidurile Bethesda pentru selectarea pacienților afectați de cancer colorectal pentru analiza MSI. Aceste ghiduri au fost revizuite și publicate în 2004 pentru a include istoricul familial și caracteristicile patologiei cancerului colorectal, precum celule în inel cu pecete, reacții de tip sindrom Crohn, caracteristici mucinoase și localizarea tumorii la nivelul colonului drept. Aceste ghiduri generale au limitări, deoarece multe familii cu sindrom Lynch nu vor întruni criteriile Amsterdam sau ghidurile Bethesda. Totodată, în ciuda îndeplinirii criteriilor sau ghidurilor, unele familii nu vor poseda alterări germinative ale oricăreia dintre genele MRR. În 2008, Hampel et al. au demonstrat fezabilitatea imunohistochimiei utilizată la scală largă, ce ar putea ajuta în direcționarea spre testare genetică. În 2009, la Jerusalem Workshop, s-a recomandat testarea MSI de rutină și imunohistochimie pentru toate cancerele colorectale diagnosticate la pacienți sub vârsta de 70 de ani. Aceste recomandări au fost incorporate în raportul Evaluării Aplicației Genomice în Clinică și Profilaxie.
Ghid clinic de diagnostic in sindromul LYNCH
Criteriile Amsterdam I
Cel puțin 3 rude cu un cancer colorectal confirmat histologic:
- Una este rudă de gradul I pentru celelalte două;
- Cel puțin 2 generații consecutiv afectate;
- Cel puțin o rudă diagnosticată cu cancer colorectal <50 ani
- Polipoza adenomatoasă familială (PAF) a fost exclusă.
Criteriile Amsterdam II
Cel puțin 3 rude cu un cancer asociat cu cancerul colorectal ereditar non-polipozic (HNPCC) [neoplasm colorectal, endometrial, gastric, ovarian, de ureter, bazinet, intestin subțire, tract hepatobiliar, piele (tumori sebaceice)]:
- Una este rudă de gradul I pentru celelalte două;
- Cel puțin două generații consecutive sunt afectate;
- Cel puțin o rudă diagnosticată cu un sindrom asociat cancerului să fie diagnosticată la o vârstă <50 ani;
- PAF exclusă în orice caz de cancer colorectal;
- Tumorile verificate oricând este posibil.
Ghidurile Bethesda revizuite
Tumorile colorectale ale unui individ trebuie testate pentru MSI în următoarele situații:
- Cancer colorectal diagnosticat la un pacient cu vârsta <50 ani împliniți.
- Prezența tumorilor colorectale sincrone, metacrone sau a oricăror altor tumori asociate HNPCC, indiferent de vârsta la care au fost diagnosticate.
- Cancer colorectal cu instabilitate crescută a microsateliților diagnosticată la un pacient cu vârsta <60 ani împliniți.
- Cancer colorectal diagnosticat la una sau mai multe rude de gradul I, diagnosticate cu tumori asociate HNPCC, dintre care una să fie diagnosticată la o vârstă <50 ani.
- Cancer colorectal diagnosticat la două sau mai multe rude de gradul I și II, diagnosticate cu tumori asociate HNPCC, indiferent de vârsta la care s-a efectuat diagnosticul.
Genetica sindromului Lynch
Majoritatea cancerelor colorectale (CCR) se dezvoltă sporadic în urma alterărilor somatice ale celulelor epiteliale din colon; cu toate acestea, în aproximativ 30% din cazuri, CCR se dezvoltă în pacienți cu un istoric familial pozitiv pronunțat.
Pacienții cu rude de gradul I afectate au un risc crescut de 2-10 ori de a dezvolta CCR iar, în absența sindroamelor polipozice sau sindromului Lynch, pacienții sunt probabil purtători ai unor variante cu penetranță incompletă într-un interval de gene.
Pacienții afectați de sindromul Lynch au moștenit cel puțin o alelă patogenă a unei gene MMR. Funcția normală a proteinelor MMR este de a verifica secvența nucleotidică și de a corecta posibilele erori ce pot apărea în timpul sintezei ADN.
Microsateliții sunt secvențe scurte, repetitive, ce sunt distribuite în întregul genom uman.
Proteinele MMR defecte produc variante în cadrul microsateliților, ce se manifestă ca și un câștig sau o pierdere în ceea ce privește lungimea repetițiilor. Acest fenomen este descris ca și instabilitatea microsateliților (MSI).
Cancerele ce posedă variații în proporție mai mare de 40% în cadrul microsateliților (ex. pozitivitate pentru 2 sau mai mult, din cei 5 markeri standard pentru microsateliți testați în mod obișnuit) sunt descrise ca având instabilitate microsatelitară cu frecvență ridicată (MSI-H). Interesant este că acest fenotip a fost observat la 15% dintre cazurile sporadice de CCR datorită metilării somatice a regiunii promotoare a genei MLH1.
Se poate efectua în continuare genotiparea pentru mutația somatică V600E a genei BRAF, pentru a confirma evenimente somatice de instabilitate microsatelitară. Mutațiile BRAF împreună cu metilări ale genei MLH1 sunt tipice pentru CCR sporadic și nu sunt regăsite la pacienții diagnosticați cu sindrom Lynch.
Tumorile ce nu au instabilitate microsatelitară sunt stabile microsatelitar (MSS), iar cele ce posedă variații în proporție de mai puțin de 40% a sateliților (ex. unul din cei 5 markeri standard ce arată instabilitate microsatelitară) prezintă instabilitate microsatelitară cu frecvență scăzută (MSI-L), cu toate că acest grup nu are o relevanță clar stabilită până în prezent și aceste tumori nu sunt considerate instabile microsatelitar.
Majoritatea indivizilor diagnosticați cu sindrom Lynch posedă cel puțin o mutație germinală patogenică a genelor MRR: MLH1, MSH2, MSH6, PMS2.
MLH1 și MSH2 sunt genele cel mai des „mutante”, mutațiile acestora fiind diagnosticate la aproximativ 70% dintre pacienții diagnosticați cu sindrom Lynch (32% – mutații ale genei MLH1 și 38% – mutații ale genei MSH2).
Pacienții purtători de mutații ale genei MSH2 au o predispoziție crescută de a dezvolta cancere extracolonice și o frecvență mai scăzută a CCR, comparativ cu cei purtători de mutații ale genei MLH1.
Mutațiile MSH6 sunt asociate în mod obișnuit cu neoplasmul gastrointestinal și endometrial, cu o vârstă de diagnostic mai târzie.
Mutațiile MSH6 au fost recunoscute ca o cauză frecventă pentru sindromul Lynch atipic (nu îndeplinește nici un criteriu Amsterdam).
Senter et al. au analizat 99 de probanzi diagnosticați cu tumori asociate sindromului Lynch ce au revelat o pierdere izolată a PMS2 și au demonstrat apariția mutațiilor germinale ale PMS2 la 62% dintre probanzi. Printre familiile cu mutații monoalelice ale PMS2, 65.5% au îndeplinit ghidurile Bethesda revizuite, iar penetranța mutațiilor monoalelice la purtători a fost mai scăzută față de purtătorii de mutații ale altor gene MMR.

Recent, delețiile 3’ constituționale ale EPCAM au fost demonstrate a fi o cauză de sindrom Lynch prin suprimarea epigenetică a MSH2 în țesuturile ce exprimă EPCAM, rezultând în deficiență de MSH2 specifică de țesut.
Kempers et al. au efectuat un studiu de cohortă, comparând 194 de pacienți purtători de deleție EPCAM și 473 de pacienți purtători de mutații ale genelor MLH1, MSH2, MSH6 sau a combinației de deleție EPCAM – MSH2. Purtătorii deleției EPCAM au prezentat un risc cumulativ de 75% de a dezvolta CCR înaintea vârstei de 70 de ani, ceea ce nu diferă semnificativ față de cea a purtătorilor combinației de deleție EPCAM-MSH2 sau de mutație a MSH2, dar a fost crescut față de cel al purtătorilor de mutație a MSH6. Doar cei ce au fost purtători de deleție ce se extindea în proximitatea regiunii promotoare a MSH2 au avut un risc crescut de apariție a cancerului endometrial. Astfel, aceste rezultate subestimează efectul deficienței MSH2 în mozaic, conducând la riscuri variabile de dezvoltare a cancerului, ceea ce ar putea forma baza pentru protocoale optimizate în privința recunoașterii și profilaxiei specifice a cancerelor la pacienții purtători de deleție EPCAM.
Până în prezent, studiile de asociere genomică (GWAS) au identificat aproximativ 20 de variante genice asociate cu dezvoltarea CCR sporadic. Wijnen et al. au identificat polimorfismul nucleotidic (SNP) rs16892766 (8q23.3) și rs3802842 (11q23.1) ca fiind asociat, semnificativ statistic, cu riscul de dezvoltare a CCR în familiile afectate de sindrom Lynch. Pentru SNP-ul rs16892766, prezența alelei C a fost asociată cu un risc crescut de dezvoltare a CCR, într-un model dependent de „doză”, astfel că homozigoții CC au avut un risc asociat de 2,16 ori mai mare. Pentru SNP-ul rs3802842, riscul elevat de dezvoltare a CCR asociat cu alela C a fost regăsit doar la purtătorii de sex feminin, în timp ce riscul de dezvoltare CCR a fost semnificativ crescut la purtătorii homozigoți în comparație cu cei heterozigoți. Printr-un model aditiv pentru ambele variante, riscul a fost semnificativ asociat cu o multitudine de alele de risc. Efectele au fost mai puternice la purtătorii de sex feminin decât la cei de sex masculin. Acești factori ar putea ajuta în identificarea indiviziilor cu risc crescut ce necesită o urmărire mai amănunțită.
În mod interesant, dintre toate familiile ce îndeplineau criteriile Amsterdam I, aproximativ 80% dintre familii poartă o anomalie ereditară a unei gene MMR. Pentru a determina dacă riscul de dezvoltare a CCR ce există în cadrul familiilor Amsterdam-1 fără deficit aparent al MMR este diferit față de familiile Amsterdam-1 cu anormalități ADN MMR prezente, Lindor et al. au identificat 161 de pedigriuri pozitive conform criteriilor Amsterdam-1 și au comparat familiile cu și fără anormalități ale MMR. Familiile ce au îndeplinit criteriile Amsterdam-1 și nu prezentau defecte ale ADN MMR nu au prezentat aceeași incidență a cancerului comparativ cu familiile cu deficiență MMR ereditară. Rudele din cadrul acestor familii prezentau un risc moderat de a dezvolta CCR la o vârstă înaintată, fără neoplazii extracolonice, față de cei ce prezentau defecte identificabile ale MMR. Astfel, pentru a discrimina cele două cohorte, s-a propus denumirea „CCR familial tip X” pentru acest tip de agregare familială a CCR.
Proiectul Variomului Uman a stabilit recent un proiect pilot, în colaborare cu Societatea Internațională a Tumorilor Gastrointestinale Ereditare (InSiGHT), pentru a investiga toate variantele transmisibile ce afectează genele susceptibile pentru cancerul de colon. Datele de tip genotip-fenotip au fost depozitate în bazele de date a variantelor genelor pentru cancerul de colon InSiGHT. Acest registru pune la dispoziție posibilitatea înțelegerii mai profunde a formelor rare, dar și comune a sindroamelor neoplazice colorectale ereditare. Recent, InSiGHT a creat o comisie internațională de cercetători și clinicieni pentru a revizui variantele genelor MMR trimise bazelor de date pentru a dezvolta, testa și aplica o schemă pe 5 etape de a clasifica cele 2.360 de variante unice constituționale ale genelor MMR. Din cele 12.006 variante introduse în baza de date, 2.641 de variante au fost incluse în Clasa 5 (patogenic), 239 în Clasa 4 (probabil patogenic), 6.982 în Clasa 3 (necunoscut), 53 în Clasa 2 (probabil non-patogenic) și 2.091 în Clasa 1 (fără rol patogenic cunoscut). Aceasta este primul efort de clasificare comprehensivă la scară largă pentru a crea o baza de date locus-specifică și de a furniza informații în legătură cu patogenitatea variantelor descoperite. Inițiativa InSiGHT a oferit un model cheie în dezvoltarea colaborării multidisciplinare pentru interpretarea transparentă a variantelor ADN. Criteriile dezvoltate oferă o fundație pentru standardizarea clasificării clinice a variantelor, ceea ce va ajuta în informarea pacientului și familiei, dar și pentru managementul pacientului, cu ajutor consilierii genetice.
Profilaxia sindromului Lynch
Sindroamele neoplazice colorectale ereditare reprezintă un spectru de patologii ce au ca și cauză un număr echivalent de mare de mutații germinale. Deși sindromul Lynch reprezintă doar 5% din cazurile de cancer colorectal, pacienții afectați poartă un risc de dezvoltare în timpul vieții a cancerului colorectal și altor neoplazii de 80%, la o vârstă medie de debut de 46 de ani. Acest fapt are implicații semnificative asupra sănătății publice, în special asupra selectării pacienților pentru programele de profilaxie.
Multiple ghiduri în ceea ce privește controlul CCR familiale și profilaxia lor au fost propuse de multiple corpuri de expertiză, cel mai mare dintre acestea fiind cel al National Comprehensive Cancer Network (NCCN). Mutațiile germinale ale genelor MMR sunt descoperite în maxim 80% din cazuri de sindrom Lynch, în ciuda îndeplinirii criteriilor Amsterdam, prezenței instabilității microsateliților cu frecvență crescută sau pierderii expresiei proteinelor MMR. Sindromul este acum un amalgam ce cuprinde o sumedenie de entități moleculare – chiar și cu introducerea unor ghiduri Bethesda actualizate, până la 28% dintre purtătorii de mutații ale genelor MMR ar fi neidentificați. Ghidurile Amsterdam și Bethesda au fost criticate pentru lipsa specificității și sensibilității, astfel că sunt dificil de aplicat în practica clinică. Alte studii au demonstrat că ghidurile Bethesda pot omite 6%-25% dintre purtătorii de mutații. Astfel, clinicienii ar trebui să facă efortul major de a documenta istoricul familial detaliat al tuturor pacienților pentru a identifica familiile afectate de sindrom Lynch.
O dezbatere s-a declanșat în jurul testării instabilității microsateliților strict prin PCR sau prin evaluarea expresiei proteinelor MMR cu ajutorul imunohistochimiei, deoarece există o strânsă concordanță între cele două. Colorațiile imunohistochimice oferă o metodă rapidă și cost-eficientă de testare a expresiei proteinelor sintetizate de genele MLH1, MSH2, MSH6 și PMS2. Dacă una sau mai multe dintre aceste colorații nu sunt pozitive, testarea genetică germinală poate fi țintită asupra acelei gene. Pentru a implementa programele de profilaxie a tumorilor sindromului Lynch sau a identifica bariere ce ar putea opri sau întârzia aceasta, National Society of Genetic Counselors Cancer Special Interest Group a condus un sondaj online în iulie 2011. Protocoale de profilaxie pentru tumorile sindromului Lynch în vederea diagnosticării cancerelor de colon și/sau de endometru au fost aprobate de 52.8% dintre respondenți, iar aproximativ jumătate dintre aceștia doreau abordarea universală. O mare varietate există în ceea ce privește metodele de screening; 64,2% ar fi început cu imunohistochimie, 20,8% ar fi început cu testarea MSI, iar 15,1% ar fi efectuat ambele pentru diagnosticul nou de CCR. Doar 21,7% dintre respondenți ar fi aprobat un program de profilaxie pentru cancerul endometrial diagnosticat de novo. Consimțământul informat a fost rareori obținut (7,1%) și a existat o lipsă a uniformității în ceea ce privește primirea rezultatelor de către pacienți. Câteva limitări legate de implementare au fost costurile, aducerea jucătorilor cheie la un consens și convingerea cadrelor medicale de necesitate acestora.
În general, programele de profilaxie a tumorilor sindromului Lynch ce există în acest moment variază mult. Algoritmul actual pentru diagnosticul sindromului Lynch include atingerea criteriilor din ghidurile Bethesda. Dacă aceste criterii nu sunt îndeplinite, unii autori consideră că nu există nici un avantaj în a continua cu testarea genetică. Pe de altă parte, alți autori susțin testarea profilactică de rutină a specimenelor patologice de CCR pentru instabilitatea microsateliților și expresia imunohistochimică a MMR. Această abordare s-a demonstrat ca având o mai bună rată de identificare a purtătorilor de mutații, comparativ cu cei ce au folosit exclusiv ghidurile Bethesda. Sindicatul Evaluării Aplicațiilor Genomice în Clinică și Profilaxie (EGAPP) au pledat pentru introducerea profilaxiei pentru sindromul Lynch la toți pacienții nou-diagnosticați cu CCR, în ideea de a identifica toate rudele la risc. Recent, ghidurile revizuite pentru managementul clinic al sindromului Lynch recomandă ca toate cancerele colorectale și endometriale cu debut sub vârsta de 70 de ani să fie testate imunohistochimic sau pentru a identifica instabilitatea microsateliților, în ideea de a identifica potențiali pacienți afectați de sindrom Lynch. Aceste recomandări au fost incorporate în ghidurile actualizate NCCN pentru Evaluarea Riscului Familial/Genetic: Versiunea Colorectală I.2014
Concluzii
Deși există imperfecțiuni în ceea ce privește testarea genetică la acest moment, raționamentul clinic ar trebui să dicteze un plan de management, iar pacienții la risc ar trebui incluși în programe de profilaxie a cancerului colorectal. Pacienții purtători confirmați ca având mutație a unei gene MMR ar trebui să efectueze o colonoscopie o dată la 1-2 ani, începând cu vârsta de 20-25 de ani, sau cel puțin la 10 ani de la cea mai mică vârstă de debut a cancerului în familie. S-a observat o creștere a numărului de programe de profilaxie a tumorilor asociate sindromului Lynch începând cu anul 2000, iar instituțiile adoptă într-un ritm alert abordarea profilaxiei universale. Este timpul pentru standardizarea instituțională a clinicilor ce tratează pacienți cu risc elevat pentru CCR sau CCR ereditar. Ghidurile internaționale ar trebui să determine criteriile și calitatea standard pentru a determina care sunt aceste clinici.
BIBLIOGRAFIE
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Clendenning, M.; Sotamaa, K.; Prior, T.; Westman, J.A.; et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J. Clin. Oncol. 2008, 26, 5783–5788.
- De Jong, A.E.; Morreau, H.; van Puijenbroek, M.; Eilers, P.H.; Wijnen, J.; Nagengast, F.M.; Griffioen, G.; Cats, A.; Menko, F.H.; Kleibeuker, J.H. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology 2004, 126, 42–48. Genes 2014, 5 504
- Thorson, A.G.; Knezetic, J.A.; Lynch, H.T. A century of progress in hereditary nonpolyposis colorectal cancer (Lynch syndrome). Dis. Colon Rectum 1999, 42, 1–9.
- Lynch, H.T.; Kimberling, W.; Albano, W.A. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). I. Clinical description of resource. Cancer 1985, 56, 934–938.
- Lynch, H.T.; Schuelke, G.S.; Kimberling, W.J. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). II. Biomarker studies. Cancer 1985, 56, 939–951.
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425.
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary
nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456.